PNF: Extension spaces for phylogenetic trees
Amy D Willis
February 7 2025
pnf_extn_spaces.RmdPublished paper | Software | Supplementary | Preprint
TL;DR: The StatDivLab has a big vision to revolutionize how you estimate bacterial, archaeal, and interdomain phylogenetic trees. This paper is our first step in this direction. This method is quite specific, and may not be feasible for your analysis, but it reflects some ideas we’re excited about and are advancing further. Stay tuned.
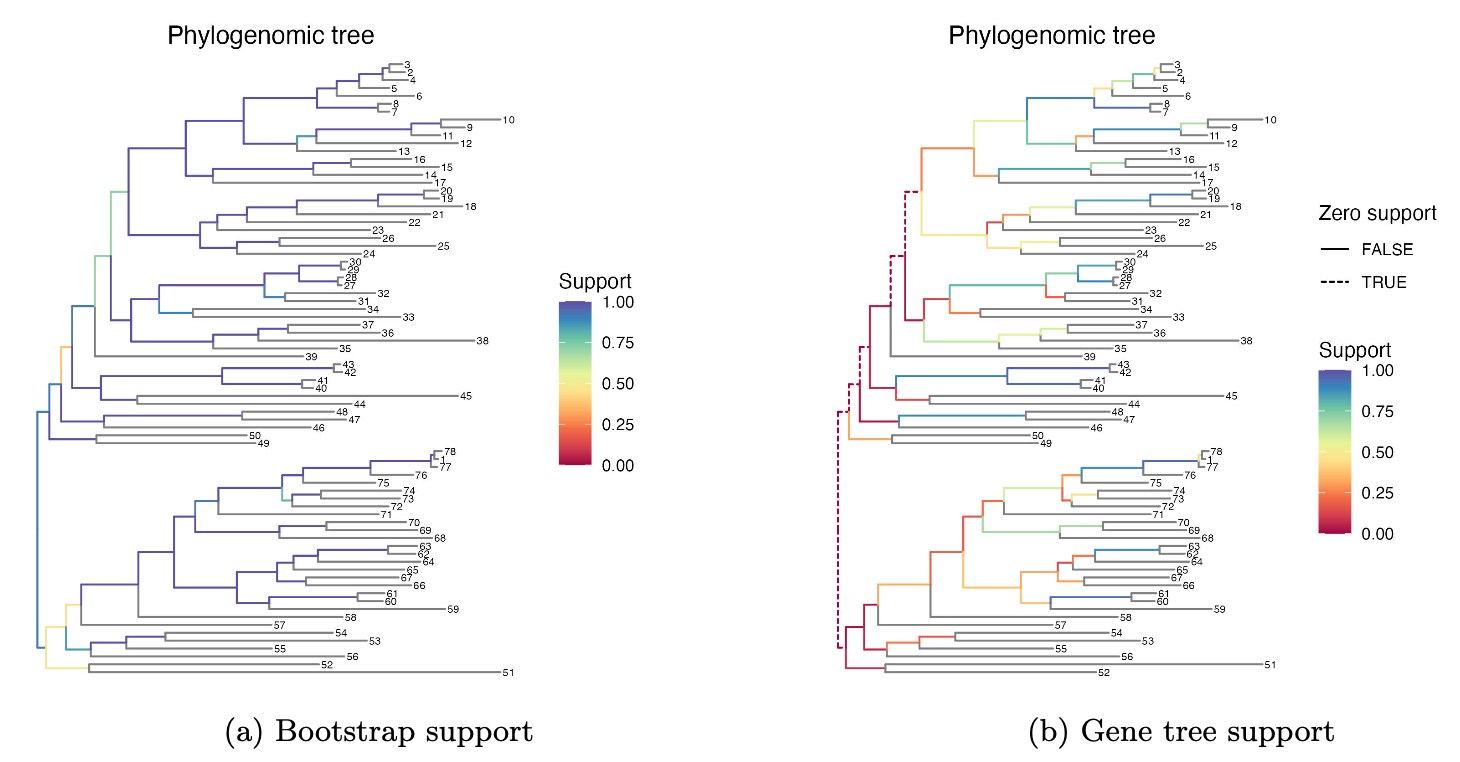
Long term plan: Better estimates of bacterial phylogenies, with accurate and clear uncertainty measures. Address this “100% bootstrap support for splits supported by no genes” nonsense. If you don’t believe me, here’s Supplementary Data Figure 7 from our “groves” paper:
This paper: A distance between phylogenetic trees with different leaf sets
Why is this important: Different genes provide complementary information about shared ancestry. I believe that while we should be selective about what genes we use to estimate phylogenies, we shouldn’t be concatenating them together and pretending it’s one supergene. Instead, we should estimate gene-level phylogenies and combine them cleverly. But, not all genomes will have all core genes (e.g., imperfect assemblies, but other reasons too), and we still care about those genomes. We therefore have a collection of phylogenetic trees to combine… but they don’t have exactly the same leaves. How do you compare trees that don’t have the same leaves? I like to compare trees that do have the same leaves using the BHV distance. That’s why we came up with an algorithm that extends the BHV distance to accommodate non-identical leaf sets.
My favourite things: Resolves (IMO) the biggest limitation of BHV tree space for analyzing bacterial gene trees. Paves the way for faster approaches. Creates the first data-analytic tools for non-identical leaf-ed trees in BHV space.
My least favourite thing: There are so many dang tree topologies that it’s not really computationally feasible in practice. Also, the geometry of extension spaces means the distance is not formally a metric (both positivity and triangle inequality RIP).
Use cases: Rather limited. Your typical microbial ecologist probably won’t use this. They might use this, though. Target audience is more likely BHV space supernerds, convex optimisation nerds, phylogenetic tree distance afficionados.
Top shoutouts: We leaned heavily on this work from Megan Owen and Gillian Grindstaff. I still love this paper on tree visualization with Sarah Teichman and Mike Lee.
Review process: Amazingly smooth. Sparkly mineral water in a desert wasteland. Reviewers were constructive, reasonable and fast. I’ll definitely submit to IEEE TCBB again.
Misc: Dr Maria Valdez was awarded the School of Public Health Outstanding PhD Award for this work. Go Maria! Thoroughly well-deserved.
Wrap-up: Stay tuned! Practical tools for bacterial phylogenetics are in the works…